Modeling admixture
We develop models and methods for reconstructing the admixture history of populations, namely the times, sources, and magnitudes of historical gene flow events. For example, individuals in admixed populations differ in the proportion of ancestry they carry from each ancestral source. This can be modeled and used for inferring the historical admixture events [1]. Another method is based on a Hidden Markov Model for the changes in ancestry along admixed genomes.
[1] Xue et al., PLoS Genetics, 2017 (link)
Modeling the effect of recombination parameters on patterns of genetic variation
Recombination is usually modeled as a simple Poisson process along the genome. However, in reality, the molecular process is more complex. We developed new theoretical results for models of crossover interference, which is a constraint on the distance between nearby crossovers. The results may allow the study of interference over multiple meioses, even when crossovers from individual meioses cannot be distinguished.
Approximations of the coalescent with recombination
Under the coalescent with recombination, the time to the most recent common ancestor at each genomic position depends on the history of the entire upstream sequence. Sequentially Markov coalescent models (SMC and SMC’) have relaxed this constraint to enable the development of extremely fast population-genetic inference methods, capable of analyzing entire genome sequences. However, theory was lacking for key properties of the Markovian models. We derived the joint distribution of tree heights under SMC’ for two fixed loci, as well as multiple other results [1].
In reality, even the full coalescent model ignores the fact that for diploid organisms, the genealogy of every pair of haplotypes is restricted by the underlying pedigree connecting the two individuals carrying them. Therefore, genealogies are correlated (though weakly) at distant or even completely unlinked sites. We showed theoretically that this correlation results in a non-zero variance of estimators of the mutation rate, even for infinitely many sites [2].
[1] Wilton et al., Genetics, 2015 (link)
[2] King et al., Theoretical Population Biology (2017) (link)
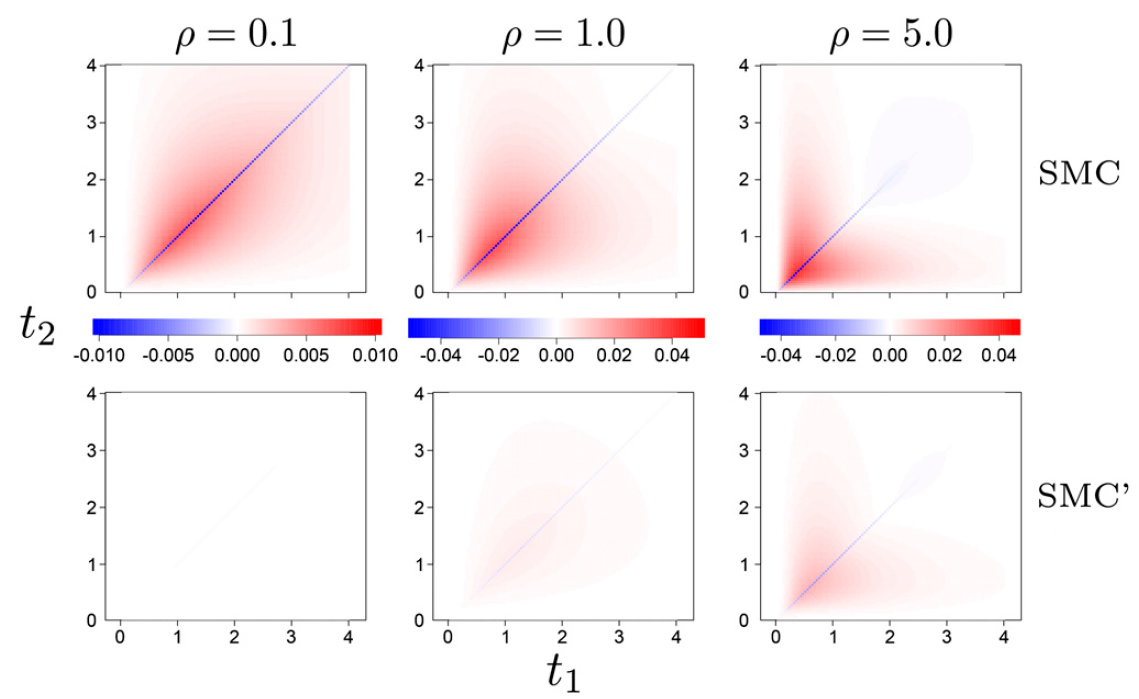
The joint density of the time to the common ancestor (t1 and t2) at two genomic loci separated by distance rho. Each panel shows the difference between the densities under the complete model (the coalescent with recombination) and one of the Markovian approximations, SMC or SMC’. From Wilton et al., Genetics, 2015.
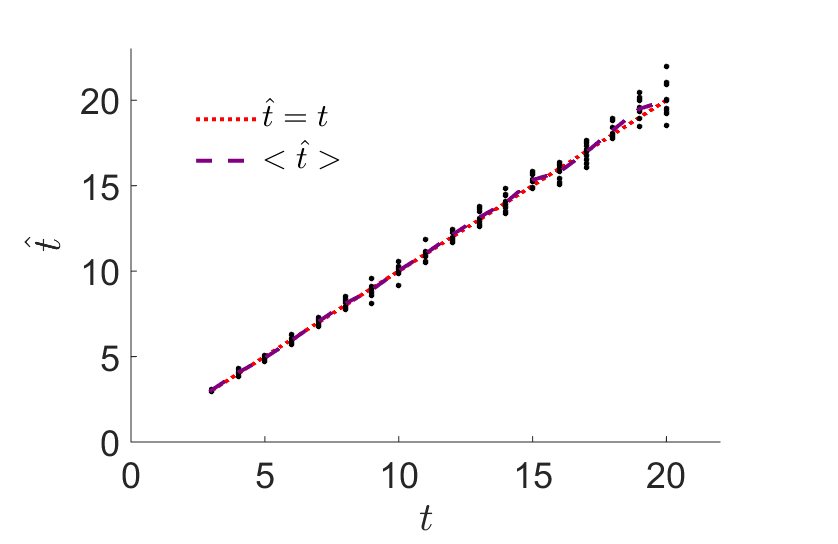
Simulation results for the inference of the time of admixture between two populations. We first recorded, for each individual, the proportion of the genome coming from each ancestral population. We then used our newly developed expression for the distribution of ancestry proportions to infer the admixture time. The inferred time is plotted vs the true, simulated time.
